TACTIC-R
TACTIC-R

Multi-Arm Therapeutic Study in Pre-ICU Patients Admitted with COVID-19
The TACTIC-R clinical trial is testing whether existing drugs, which are already used to treat other immune-related conditions, can prevent the development of severe symptoms in patients hospitalised with confirmed COVID-19. This approach, known as repurposing, is a way of finding out whether existing therapies, which are likely to be readily available in hospitals, are effective in treating the disease.
The treatments currently being tested in TACTIC-R are:
- Baricitinib - used to treat severe rheumatoid arthritis
- Ravulizumab - used to treat blood diseases where the immune system destroys red blood cells
Important Update: Remdesivir and TACTIC-R
A limited supply of Remdesivir is now available nationally to patients with COVID-19 related diseases.
- Treatment of patients with Remdesivir is NOT a contraindication to participation in TACTIC-R
- Participants should be treated with Remdesivir as standard of care whilst participating in TACTIC-R
- All sites are encouraged to use Remdesivir whenever the patient is eligible
- Use of Remdesivir will be captured as a concomitant medication during the study
The criteria for the use of Remdesivir are listed below:
Remdesivir and TACTIC-R Criteria for Use
COVID-19 Therapeutic Alert: EAMS for Remdesivir in the Treatment of COVID-19
Welcome to the patients area for the TACTIC-R trial. This trial is recruiting people who have been hospitalised with confirmed COVID-19 disease. The purpose of this trial is to prevent organ damage and reduce the need to transfer patients to ICU and ventilation.
The following provide answers to questions you might have about this trial:
Patient Information Sheet and Informed Consent Form (Short Version)
Patient Information Sheet and Informed Consent Form (Full Version)
- What is the purpose of the trial?
-
COVID-19 was declared a pandemic on 11th March 2020 by the World Health Organisation (WHO). It is a disease affecting the lungs, and is caused by a new coronavirus known as SARS-CoV2.
Some people with the virus have mild symptoms. However, some people, for example older adults and those with underlying health conditions including heart disease and diabetes, may develop more severe symptoms, leading to hospitalisation, increased support for breathing (e.g. use of a ventilator in an intensive care unit) or even death. There are currently no vaccines against the virus, or proven treatments to help combat it.
Severe symptoms of the virus are thought to be a result of an exaggerated immune response, leading to organ damage in the body. The purpose of this trial is to determine the best way to treat patients with COVID-19 infection by comparing different treatments which act on the immune system, with the aim of reducing severe symptoms and therefore the number of Intensive Care Unit (ICU) admissions in hospital.
- What is the drug being tested?
-
This trial is known as a platform study. This type of study looks at several different treatments for one same condition. As the study progresses, additional drug treatment arms can be added or removed from the study or dosing can change.
If the treatment arm you have been assigned to is stopped, you will immediately stop treatment. Once you have consented, you will be treated with any one of the following arms:
- Baricitinib - used to treat severe rheumatoid arthritis
- Ravulizumab - used to treat blood diseases where the immune system destroys red blood cells
- Standard of care - this is any treatment you would have had if you had not joined in this trial
- Who is being invited to take part?
-
Patients may be invited to participate in this trial if they are or are suspected to be COVID-19 positive, are considered to be at moderate risk of developing severe symptoms and where it is believed that baricitinib or ravulizumab may be suitable treatments.
We plan to include 1167 participants with COVID-19 disease from up to 15 hospitals across the UK.
- Do I have to take part?
-
No, participating in this trial is completely voluntary. If you do decide to participate you will be asked to sign an Informed Consent Form, however you are still free to change your mind and leave the trial at any time without giving a reason. If you choose not to participate or to leave the trial, your future medical treatment will not be affected in any way.
- What will happen to me if I take part?
-
The following flowchart summarises the trial:
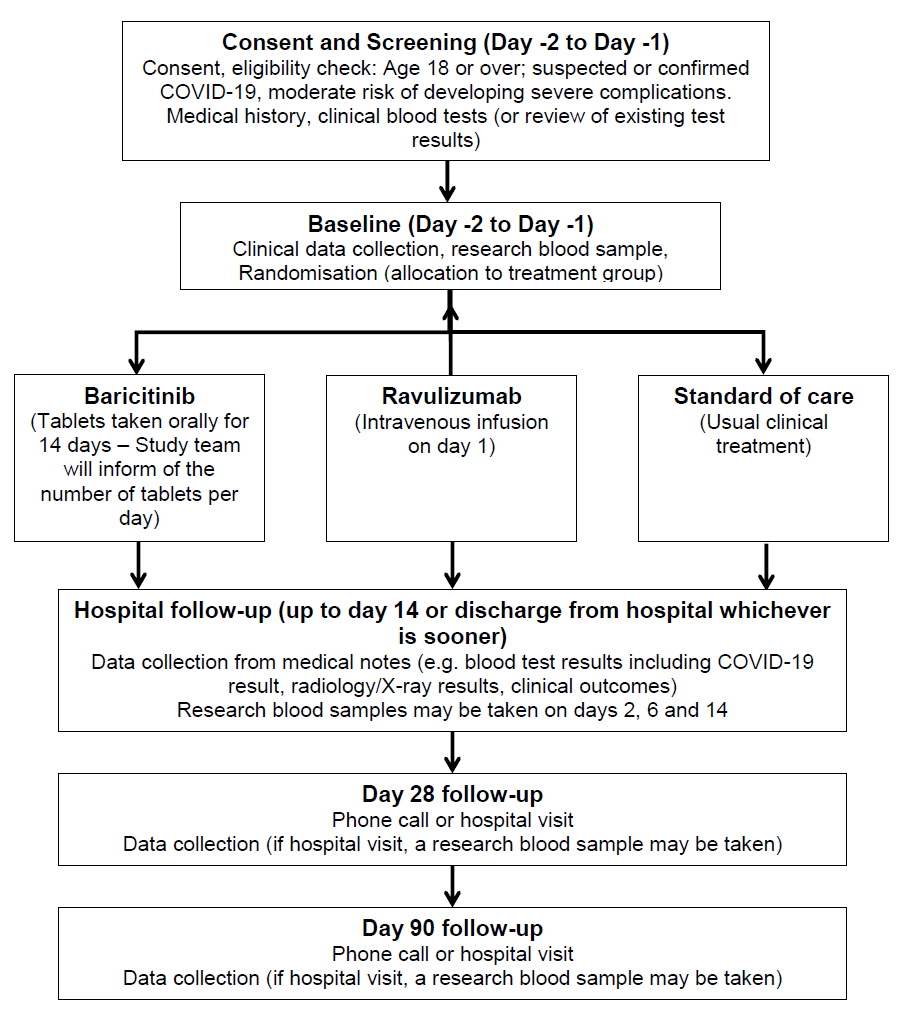
As we don’t know whether baricitinib or ravulizumab can be used to treat COVID-19 related disease, we need to compare the treatments separately to regular standard of care. There are 3 groups in this study and each group will receive a different treatment. The trial treatments mentioned above will be in addition to your usual clinical care.
Please note, all assessments up to day 14 are conducted at the hospital either up to day 14 or discharge from hospital whichever is sooner.
Consent and screening (approximately 45 minutes): If you agree to participate in the trial, you will sign the Informed Consent Form at the end of this document and be given a copy of this to take away and refer to later. The screening assessments include checking your clinical and medication history, checking that you are suitable for the trial, and reviewing blood tests you have had since admission to hospital. Where the blood tests have not yet been done, these will be taken and checked. The screening process may happen up to 48 hours before trial treatment begins.
However, since COVID-19 requires urgent treatment, where urgency is needed, screening and baseline procedures can happen on the same day. Where you are able to understand but unable to physically sign the consent form, either a family member or independent doctor can act as a witness and sign the consent form to attest that the information was accurately explained to, and apparently understood by you.
Baseline (duration, approximately 45 minutes): Further routine clinical information from your medical notes will be collected. You will also be invited to give a blood sample (blood group, serum and DNA/genetics sample for future analysis; the maximum amount of blood taken will be approximately 30ml [2 tablespoons]). If you are suitable for the trial, you will be randomised to a treatment group.
Randomisation: Brief details identifying you and answering a few questions about your health and medical conditions will be entered into a computer. The computer will then allocate you at random (like rolling a dice) to one of the possible treatment options with a chance of 1:1:1. This means you have equal chances of being assigned to one of the arms. In all cases this will include the usual standard of care for your hospital.
Treatments and in-hospital follow-ups (days 1-14, or up to discharge from hospital, whichever is sooner): Following random allocation to a treatment group, treatment will commence. The options are:
- Baricitinib (a drug which acts on certain chemicals of the immune system, and commonly used to treat rheumatological conditions)
- Ravulizumab (a drug which targets a certain pathway in the immune system, and commonly used to treat a condition known as paroxysmal nocturnal haemoglobinuria)
- Standard of care (usual clinical care for COVID infection)
We will collect information from your medical notes about how you are doing, what blood tests and x-rays/scans you have had during your hospital stay, and the results of these. On days 2, 6 and 14, research blood samples may also be taken, to look at immune cells and markers in the blood. We will take a maximum of 30ml of blood (2 tablespoons).
Day 28 and day 90 Follow-up (Telephone call or hospital visit): We will be asking a few questions about your health and we might ask you to get some bloods tests done in your local GP or at the hospital if your visit is conducted at the hospital site you were treated for your COVID infection. On a hospital visit, we may also take a further research blood sample. - What will I have to do?
-
This trial will run during your stay in hospital, therefore you will not have to move to another ward. Depending on the treatment group you are allocated to, the drugs have different administrations. These are described briefly in the trial flowchart and in more detail below:
Baricitinib
This drug is taken as tablets taken orally for 14 days; the trial doctor will tell you how many tablets you will be taking each day. Each dose should be taken with a meal or glass of water and tablets swallowed whole or crushed if you are not able to swallow.
Ravulizumab
This drug is given as an intravenous (IV) infusion (i.e. by a drip into a vein in your arm), administered at your bedside by a qualified nurse/practitioner. You will receive 1 IV infusion of this drug for this trial. If you are randomised to this arm, you will need to have a vaccine and also take some protective antibiotics before and after the vaccination.
You should tell the trial team if you feel unwell or different in any way. If you have any major concerns or are feeling very unwell please contact your trial doctor immediately using the contact numbers at the end of this information sheet.
Following discharge from hospital, we will ask you to attend or be available to talk on the phone for your day 28 and day 90 follow-up visits. If these visits happen at the hospital we may ask for further clinical and research blood samples.
You will need to carry alert card for the duration of the trial and for 8 months after completion of the trial last treatment if randomised to Ravulizumab.
You should not participate in this trial if you are planning to become pregnant or father a child during the trial. Women who are able to have a baby must use one of the following reliable forms of contraception for the entire duration of the trial and for 8 months after (if randomised to the Ravulizumab arm only) upon completion of the last treatment.
This includes:
- (1) Intrauterine Device (IUD)
- Hormonal based contraception (pill, contraceptive injection or implant etc.)
- Barrier contraception (condom and occlusive cap e.g. diaphragm or cervical cap with spermicide)
- True abstinence (where this is in accordance with the participants’ preferred and usual lifestyle)
Men are required to use adequate contraception for the entire duration of the trial and for 8 months after (if randomised to the Ravulizumab arm only) upon completion of the last treatment. This includes:
- Barrier contraception (condom and spermicide) even if female partner(s) are using another method of contraception or are already pregnant (also to protect male partners from exposure to the trial IMPs etc.)
- True abstinence (where this is in accordance with the participants’ preferred and usual lifestyle)
If you or your partner become pregnant during the trial or within 8 months (if randomised to the Ravulizumab) after completion of the last treatment, you should inform your trial doctor immediately.
You should discuss your participation in this trial with any insurance provider you have (e.g. protection insurance, life insurance, income protection, critical illness cover and private medical insurance) and seek advice if necessary, as failure to notify them may affect or invalidate your cover. - What are the side effects of the drugs?
-
Baricitinib:
Very common (more than 10% of patients): upper respiratory tract infections (including colds), high cholesterol.
Common (less than 10% of patients): nausea, cold sores, shingles, skin rash, increased risk of blood clots, sick stomach, urinary infection, pneumonia, high levels of liver enzymes.
Uncommon (less than 1% of patients): weight gain, low levels of blood immune cells, blood clots, facial swelling, acne.
Ravulizumab:
Very common (more than 10% of patients): upper respiratory tract infections, headache, common cold.
Common (less than 10% of patients): meningococcal infection, dizziness, nausea, vomiting, skin rash, itching, back, joint and muscle pain, muscle spasms, fatigue, flu-like illness, fever, chills.
- What are the possible disadvantages and risks of taking part?
-
Blood tests: Occasionally, some bruising or inflammation at the needle site may occur and very rarely infection at the puncture site.
Drug treatment: When treatments are administered by IV, you might feel slight discomfort at the site of injection. Both Baricitinib and Ravulizumab are commonly given to patients in the UK and are well tolerated. However, individual patients can react differently to the same drug which means that there is a possibility of experiencing side effects. The trial doctor will monitor any side effects regularly and take appropriate actions where necessary. If you are randomised to the ravulizumab arm, you will need to receive a meningitis vaccine when you finish your treatment. Apart from this, you will also need to take antibiotics continuously until you are vaccinated, and for a further 2 weeks after the vaccine has been given.
- What are the possible benefits of taking part?
-
There is no guarantee that you will benefit from taking part in this trial. However, information collected as part of your participation in this trial may benefit patients with COVID-19 in the future.
- What are the alternatives for treatment?
-
There are no known preventative drugs for COVID-19 currently.
- What happens when the trial stops?
-
Once the trial has ended you will be referred back to regular treatments. Pending the results of the trial, treatment guidelines may change.
- Will I receive any expenses or payment?
-
You will not receive any payment for participating in this trial and we are unable to reimburse any expenses incurred by your participation in this trial.
- What if new information becomes available?
-
Sometimes during the course of a trial, new information becomes available which might affect your decision to continue participating in this trial. Your trial doctor will contact you to discuss the new information and whether you wish to continue participating in the trial. If you still wish to continue on the trial, you will be asked to sign a new Informed Consent Form. The trial sponsor, the regulatory authority or the trial doctor may decide to stop the trial at any time. If that happens we will tell you why the trial has been stopped and arrange for appropriate care for you.
- What if I decide I no longer wish to participate in the trial?
-
You are free to come off this trial at any time without giving a reason and without affecting your future care. If you decide not to participate any further, you will no longer receive the trial treatment. No further tests will be performed on you and no further research samples will be collected. Any data already collected or results from tests already performed on you or your samples will continue to be used in the trial analysis.
The trial doctor may also choose to withdraw you from the trial if they feel it is in your best interests or if you have been unable to comply with the requirements of the trial. Reasons for trial withdrawal could include:
- You have experienced a serious side effect
- You are unable to complete the visits, medication or trial documentation as required
- The trial doctor feels you no longer appear to benefit from the treatment
If you have experienced any serious side effects during the course of the trial which require you to withdraw from the trial, your trial doctor will follow-up with you regarding your progress until the side effect has stabilised or resolved.
- What if there is a problem?
-
Any complaint about the way you have been dealt with during the trial or any possible harm you might suffer will be addressed. If you have any concerns about any aspect of this trial you should speak to your trial doctor who will do their best to answer your questions.
In the event that something does go wrong and you are harmed by taking part in the research and this is due to someone’s negligence then you may have grounds for a legal action for compensation against Cambridge University Hospitals NHS Foundation Trust (or your hospital – for multicentre trials). If your claim is successful, your legal costs will be met. The normal National Health Service complaints mechanisms will still be available to you (if appropriate).
The NHS does not provide no-fault compensation i.e. for non-negligent harm, and NHS bodies are unable to agree in advance to pay compensation for non-negligent harm. They are able to consider an ex-gratia payment in the case of a claim.
If you wish to complain or have any concerns about any aspect of the way you have been approached or treated during this trial, you can do this through the NHS complaints procedure. In the first instance it may be helpful to contact the Patient Advice and Liaison Service (PALS)) at your hospital.
- Will my taking part in this trial be kept confidential?
-
For participants recruited at Cambridge University Hospitals (where the Sponsor is also the site):
Cambridge University Hospitals NHS Foundation Trust (CUH) is the Sponsor for this clinical trial based in the UK. They will be using information from you and your medical records in order to undertake this trial and will act as the data controller for this trial. This means that they are responsible for looking after your information and using it properly. The Sponsor organisation will keep identifiable information about you for 5 years after the trial has finished to ensure your safety and allow the trial to be reviewed by the authorities after it is finished.
Your rights to access, change or move your information are limited, as the Sponsor organisation needs to manage your information in specific ways in order for the research to be reliable and accurate. To safeguard your rights, we will use the minimum personally-identifiable information possible.
You can find out more about how the Sponsor uses your information using the information below:
For Cambridge University Hospitals NHS Foundation Trust, please visit: https://www.cuh.nhs.uk/corporate-information/about-us/our-responsibilities/looking-after-your- information, or email the Data Protection Officer at: [email protected]
Cambridge University Hospitals will collect your name and contact details to contact you about this trial, and make sure that relevant information about the trial is recorded for your care, and to oversee the quality of the trial. Individuals from the Sponsor and regulatory organisations may look at your research records to check the accuracy of this trial. Cambridge University Hospitals will pass these details to the Sponsor along with the information collected from you and your medical records. The only people in the Sponsor organisation who will have access to information that identifies you will be people who need to contact you in relation to this trial and to audit the data collection process. Cambridge University Hospitals will keep identifiable information about you from this trial for 5 years after the trial has finished.
For participants recruited at other participating sites:
Your hospital will keep your name, (NHS number) and contact details to contact you about this trial, and make sure that relevant information about the trial is recorded for your care, and to oversee the quality of the trial. Certain individuals from the Sponsor(s) and regulatory organisations may look at your medical and research records to check the accuracy of this trial. The Sponsor(s) will only receive information without any identifying information.
All information collected about you as a result of your participation in the trial will be kept strictly confidential. Your personal and medical information will be kept in a secured file and be treated in the strictest confidence.
Once you have agreed to participate in this trial you will be allocated a Trial ID Number. This is a unique trial number which will be used on all your trial documentation along with your date of birth. Your date of birth is considered to be personal information. We collect this personal information on trial documentation to help ensure that the data we receive as part of your trial participation is correctly allocated to you. By cross checking these two unique references we can ensure the integrity of the data.
The people who analyse the information will not be able to identify you and will not be able to find out your name, or contact details. Only anonymous trial data, without any personal information will be published at the end of the trial.
When you agree to take part in this trial, the information about your health and care may be provided to researchers running other research studies in this organisation and in other organisations. These organisations may be universities, NHS organisations or companies involved in health and care research in this country or abroad. Your coded trial data may be sent to other country(ies) outside the European Economic Area (EEA) for analyses, where the data protection laws are not the same. .
This information will not identify you and will not be combined with other information in a way that could identify you. The information will only be used for the purpose of health and care research and cannot be used to contact you or to affect your care. It will not be used to make decisions about future services available to you, such as insurance. If you have taken part in a parallel biomarker COVID19 study at your hospital, we may wish to exchange information from the TACTIC-R trial with these study doctors to further enhance our knowledge of how the immune system handles COVID19 and responds to different treatments. All your details will be anonymised
We will need to inform your GP of your participation in this trial so that any medical decisions made by your GP account for any treatment you are receiving as part of this trial. Your GP may also contact us if they have any concerns about your participation in this study.
- What will happen to my samples?
-
Blood and serum samples that are collected in this trial will be securely stored at the site for the duration of the trial and will be accessible to authorised trial staff only. These will be subsequently transported in batches for collation and analysis to a central laboratory in Cambridge. With your permission any unused samples at the end of this trial will be stored for future tests related to this study, and for future approved research projects.
- What about genetic tests?
-
As part of the study, we may extract DNA from your sample. DNA is the chemical that makes up genes, influencing the factors we inherit and which determine our characteristics. We will also isolate and test other components of your blood such as RNA and protein and measure chemicals in the blood. We hope the results of this profiling will help us understand COVID-19 better. As this research is exploratory, you will not receive feedback regarding any ‘markers’ identified in your DNA.
- What will happen to the results of the trial?
-
The results of the trial will be anonymous and you will not be able to be identified from any of the data produced. When the results of this trial are available, they may be published in peer reviewed medical journals and used for medical presentations and conferences. They will also be published on the EU Clinical Trials Register website, a central registry for all clinical trials conducted in the EU.
Anonymous datasets from the trial may also be made available to other researchers in line with national and international data transparency initiatives.
If you would like to obtain a copy of the published results, please contact your trial doctor directly who will be able to arrange this for you.
- Who is funding the trial?
-
The trial is being funded by Eli Lilly and Company UK Ltd and Alexion Pharma UK Ltd.
- Who has reviewed this trial?
-
All research within the NHS is reviewed by an independent group of people called a Research Ethics Committee, to protect your interests. This trial has been reviewed and given favourable opinion by Cambridge Central Research Ethics Committee. The Medicines and Healthcare Products Regulatory Agency (MHRA) who are responsible for regulating medicines in the UK have also reviewed this trial.
Welcome to the staff area for the TACTIC-R trial. Adult COVID-19 patients may be invited to take part in this trial if they meet criteria for having a high likelihood of benefit from immunomodulation. A risk count has been developed to assist with the identification of patients admitted with a clinical/laboratory diagnosis of COVID19 who are at increased risk of developing severe COVID19-related disease. A video with further information about the scoring system for patient selection is available in the Training area.
Eligible patients will be randomised using a central web-based randomisation service to one of three arms initially: baricitinib, ravulizumab or usual standard of care.
- Protocol and Poster
- Patient Information Sheet and Informed Consent Form
-
Patient Information Sheet and Informed Consent Form (Short Version)
Patient Information Sheet and Informed Consent Form (Full Version)
Endothelial Cell Collection Patient Information Sheet and Informed Consent Form
- Unlocalised Site Documents
- Remdesivir and TACTIC-R
-
Remdesivir and TACTIC-R Criteria for Use
COVID-19 Therapeutic Alert: EAMS for Remdesivir in the Treatment of COVID-19
- Additional Patient Facing Documents
-
Information for Legal Representatives
TACTIC-R Pregnant Partner GP Letter for Ravulizumab
TACTIC-R Pregnant Partner GP Letter for Baricitinib
TACTIC-R Pregnant Partner PISICF
TACTIC-R GP Letter for Ravulizumab
TACTIC-R GP Letter for Baricitinib
- Substantial Amendments
-
The following amendments have been made to the trial. These folders contain the complete document set as submitted and approved:
The following videos for site staff explain key aspects of the patient recruitment process. Once you have finished the online training, please complete and submit this form to update the trial team. You will receive an emailed copy of this form.
Training Videos
The following groups and committees are responsible for overseeing the TACTIC-R trial from design to completion:
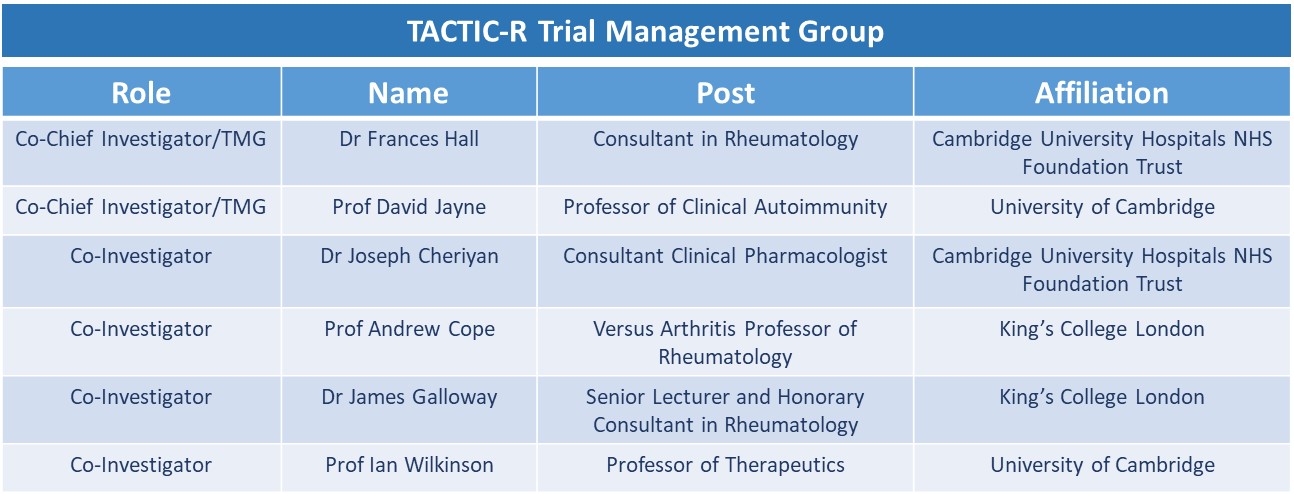


Chief Investigators
Dr Frances Hall and Professor David Jayne
Funders and Sponsors
Sponsor: Cambridge University Hospitals NHS Foundation Trust
Funders: Eli Lilly and Company and Alexion Pharmaceuticals
Key Contacts
Clinical Trial Coordinator: Elena Hernan Sancho
Telephone: 01223 349132 | Email: [email protected]


